Nonnegative matrix factorization (NMF) has been successfully applied to extract mutational signatures underlying tumor development and progression. Alexandrov L.B. and colleagures used brunet algorithm to develop a mutational signature deciphering framework. This framework has been successfully applied to 7042 cancers and extracted more than 20 signatures. However, the performance and accuracy of the brunet algorithm has not been compared with other NMF algorithms such as als, mult, sNMFR and gdclsNMF etc.
In this report, I extended this mutational signature analysis framework and conducted systematic comparison among different NMF algorithms. The result showed that brunet is worse among other algorithms in terms of reconstruction errors and stabilities. Moreover, brunet fluctuated more frequently as compared with the others. The other four algorithms except brunet have comparable performance with varying computational time spent. The sNMFR took the most time during the evaluation process. Herein, I would recommend users to the als algorithm with respect to its performance and time cost.
Mutational signature deciphering framework: https://github.com/lixiangchun/decipherMutationalSignatures.
Table 1. Evalutating the stability of different NMF algorithms on exomic somatic mutations of 544 gastric cancers
| Rank | als | brunet | gdclsNMF | mult | sNMFR |
|---|---|---|---|---|---|
| 1 | 1 | 1 | 1 | 1 | 1 |
| 2 | 0.997 | 0.999 | 0.999 | 0.998 | 0.998 |
| 3 | 0.996 | 0.613 | 0.997 | 0.996 | 0.997 |
| 4 | 0.987 | 0.72 | 0.998 | 0.99 | 0.995 |
| 5 | 0.982 | 0.725 | 0.998 | 0.988 | 0.992 |
| 6 | 0.978 | 0.906 | 0.993 | 0.969 | 0.988 |
| 7 | 0.891 | 0.969 | 0.714 | 0.792 | 0.858 |
| 8 | 0.58 | 0.827 | 0.628 | 0.742 | 0.784 |
| 9 | 0.743 | 0.734 | 0.649 | 0.631 | 0.573 |
| 10 | 0.8 | 0.585 | 0.623 | 0.461 | 0.704 |
| 11 | 0.708 | 0.525 | 0.755 | 0.389 | 0.669 |
| 12 | 0.811 | 0.512 | 0.804 | 0.216 | 0.723 |
| 13 | 0.733 | 0.532 | 0.802 | 0.31 | 0.696 |
| 14 | 0.838 | 0.431 | 0.793 | 0.315 | 0.577 |
| 15 | 0.721 | 0.376 | 0.743 | 0.256 | 0.533 |
| 16 | 0.636 | 0.446 | 0.726 | 0.156 | 0.514 |
Figure 1. Stability comparison among different NMF algorithms.
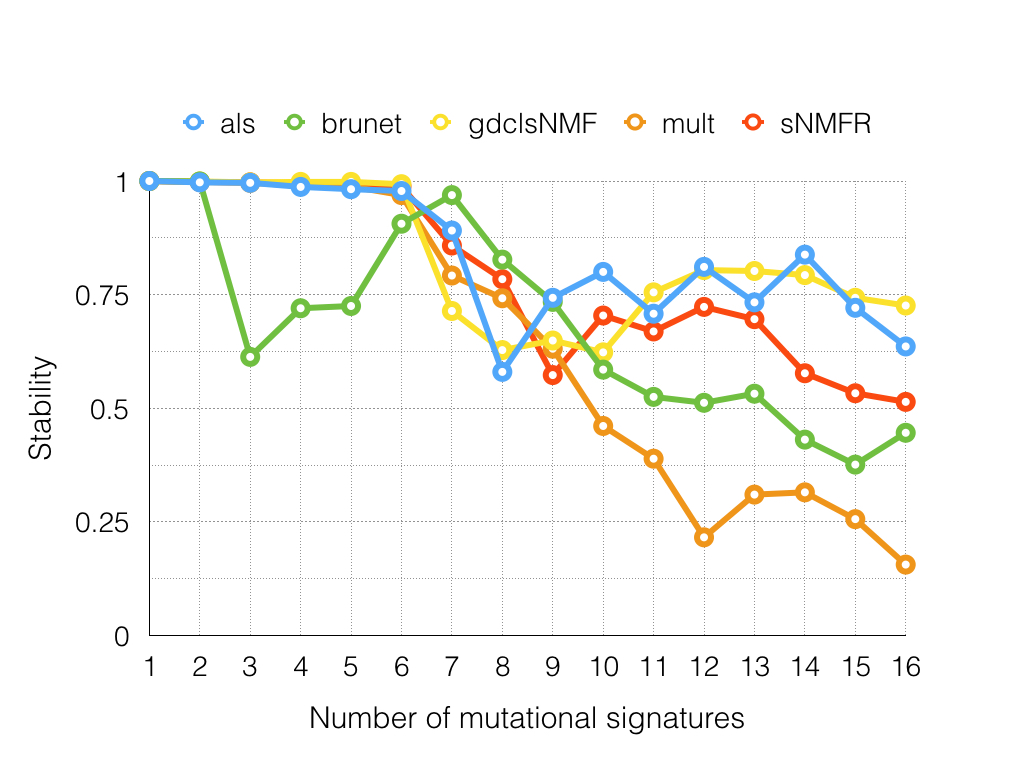
Table 2. Evalutating the stability of different NMF algorithms on exomic somatic mutations of 544 gastric cancers
| Rank | als | brunet | gdclsNMF | mult | sNMFR |
|---|---|---|---|---|---|
| 1 | 2391.52 | 2725.58 | 2395.03 | 2391.52 | 2391.52 |
| 2 | 1665.58 | 2486.25 | 1669.83 | 1666.31 | 1665.53 |
| 3 | 1222.22 | 2293.07 | 1229.19 | 1221.02 | 1219.99 |
| 4 | 852.56 | 1519.6 | 858.2 | 852.87 | 851.89 |
| 5 | 609.42 | 1237.08 | 615.44 | 609.22 | 607.98 |
| 6 | 474.87 | 936.32 | 480.04 | 474.48 | 472.06 |
| 7 | 456.48 | 608.06 | 618.83 | 469.46 | 455.93 |
| 8 | 728.42 | 580.3 | 1007.36 | 449.5 | 457.56 |
| 9 | 444.92 | 623.06 | 874.52 | 454.74 | 912.4 |
| 10 | 406.86 | 565.14 | 975.22 | 502.89 | 485.24 |
| 11 | 449.86 | 934.81 | 498.28 | 595.65 | 514.47 |
| 12 | 350.26 | 665.88 | 460.96 | 819.98 | 428.16 |
| 13 | 405.41 | 748.19 | 527.29 | 708.96 | 434 |
| 14 | 319.53 | 926.25 | 454.87 | 663.93 | 733.23 |
| 15 | 352.77 | 1077.31 | 374.9 | 751.15 | 965.44 |
| 16 | 407.35 | 677.27 | 370.51 | 754.5 | 734.07 |
Figure 2. Reconstruction error comparison among different NMF algorithms.
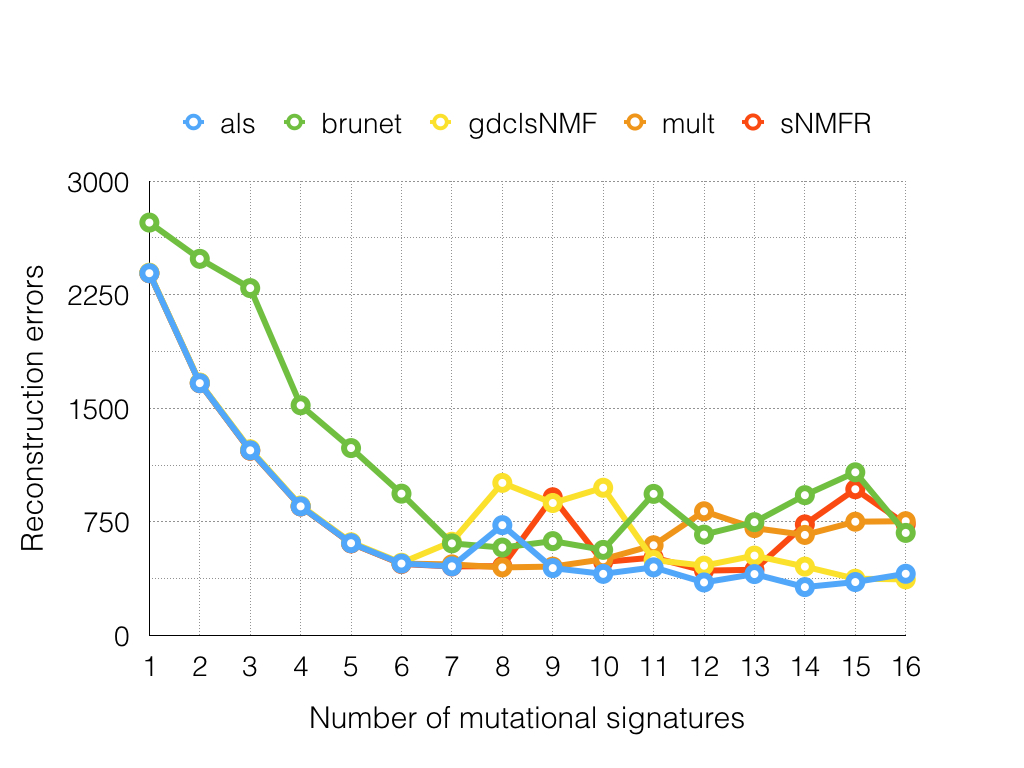
Table 3. Evaluating time cost of different NMF algorithms on exomic somatic mutations of 544 gastric cancers. Time present in hour.
| Time | als | brunet | gdclsNMF | mult | sNMFR |
|---|---|---|---|---|---|
| real | 33.1 | 2.4 | 32.9 | 63.2 | 227.4 |
| user | 42.8 | 9.7 | 54 | 191.5 | 252.5 |
| sys | 14.5 | 1.4 | 1.5 | 65.9 | 4.98 |
Stability comparison among 5 NMF algorithms
Reconstruction error comparison among 5 NMF algorithms
Mutational signature 1
Mutational signature 2
Mutational signature 3
Mutational signature 4
Mutational signature 5
Mutational signature 6
Mutational signature 7
Mutational signature 1
Mutational signature 2
Mutational signature 3
Mutational signature 4
Mutational signature 5
Mutational signature 6
All input files are listed and can be downloaded header: data.tar.gz.
-
Types file = types
-
Subtypes file = subtypes
-
Sample name file = sampleNames
-
Mutational context by sample file = originalGenomes
-
Shell script file = als.sh
#!/bin/bash
# Mutation types
typesFile=../../types
# Mutational categories for mutation types in `typesFile`
subtypesFile=../../subtypes
# Sample names, i.e. column names of input matrix in `originalGenomesFile`
sampleNamesFile=../../sampleNames
# Rows represent mutation type and columns sample names. Each cell entry
#+is the number of corresponding mutation types in the sample. Note that
#+the row number of this file must be equal row numbers of both `typesFile`
#+and `subtypesFile`.
originalGenomesFile=../../originalGenomes
# removes weak mutation types, i.e. reduces the dimmensions, default value
#+in the original code is 0.01, users can set it to 0.0 if they want to use
#+this framework as consensus clustering for gene expression analysis.
removeWeakMutationTypes=0.01
# Algorithm to use, i.e. 1. brunet, 2. mult, 3. als, 4. gdclsNMF, 5. sNMFR
# The original framework uses `brunet`.
algorithm='als'
# The min number of signatures
minNumberOfSignature=1
# The max number of signatures
maxNumberOfSignature=16
# Number of iterations to run nmf for each rank
iterationsPerCore=100
# Number of processes to use, if >1 a matlabpool is open for parallel computing.
numberOfWorkers=1
## Set to larger number is recommended, e.g. 500000
maxIterPerNmfRun=5000
# The following 2 variables must be set appropriately.
DECIPHER_MUTATIONAL_SIGNATURE_PATH=/ifshk1/BC_CANCER/03user/lixiangchun/iCGA/v0.02/decipherMutationalSignatures
MCRroot=/ifshk1/BC_CANCER/03user/lixiangchun/Software/INSTALL/MCR_R2013a/INSTALL/v81
time -p bash $DECIPHER_MUTATIONAL_SIGNATURE_PATH/run_decipherMutationalSignatures.sh $MCRroot \
$typesFile $subtypesFile $sampleNamesFile $originalGenomesFile $removeWeakMutationTypes $algorithm \
$minNumberOfSignature $maxNumberOfSignature $iterationsPerCore $numberOfWorkers $maxIterPerNmfRun